
The study followed the Declaration of Helsinki and was approved by the local ethical committee in Lund (472/2005; 2014/904; 2017/89; 2018/63). Written and oral informed consent was given by each patient.
Study cohort
A total of 219 human carotid plaques (147 plaques from ND patients and 72 from T2D patients; Supplementary Table 8) obtained from the Carotid Plaque Imaging Project biobank (Malmö, Sweden; ClinicalTrials.gov ID NCT05821894) were studied. Three patients, all asymptomatic without diabetes, underwent a second endarterectomy. The indications for surgery were plaques associated with ipsilateral symptoms (transitory ischemic attack, stroke or amaurosis fugax) and >70% stenosis measured by ultrasound, or plaques from patients with no symptoms but stenosis >80%. All patients were preoperatively examined by a neurologist. Cardiovascular risk factors, namely hypertension (systolic blood pressure >140 mm Hg), diabetes and current smoking and use of medications (anti-hypertensive drugs, diabetes treatment and statins) were recorded. Blood samples were collected prior to surgery and fasting lipoproteins (total cholesterol, HDL cholesterol, LDL cholesterol and triglycerides), HbA1c, C-reactive protein were measured. Sex (identified by the Swedish personal number) was included as a clinical variable in the study design. Reported results were, whenever possible, adjusted for sex.
Sample preparation and histology
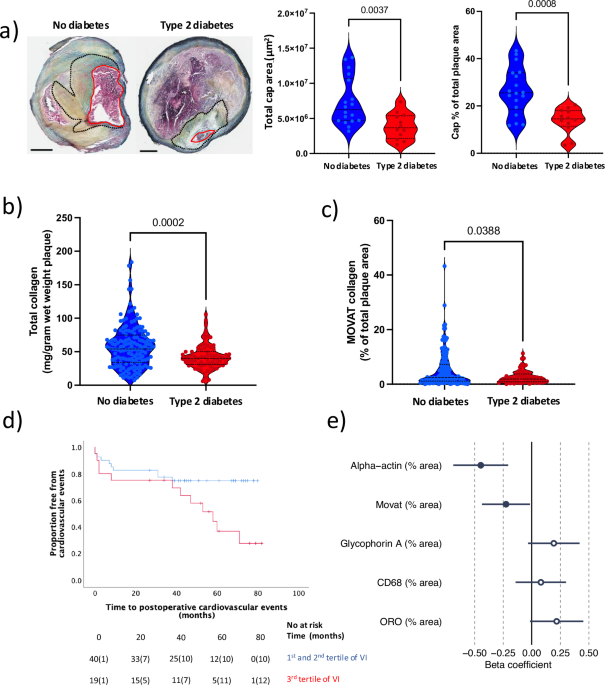
For histology and bulk RNA sequencing, plaques were snap-frozen in liquid nitrogen immediately after surgical removal, and two 1 mm fragments from the most stenotic region were kept for analysis. The remaining tissue was homogenized in 5 mL of a homogenization buffer consisting of 2 mmol/L tris(2-carboxyethyl)phosphine HCl, 1 mmol/L benzamidine, 1 mmol/L Na-orthovanadate, 10 mmol/L Na-glycerophosphate, 50 mmol/L Tris-HCl (pH 7.5), 0.25 mol/L sucrose, 50 mmol/L NaF, 5 mmol/L Na-pyrophosphate, protease inhibitor cocktail (Roche Complete, EDTA-free), and 10 mmol/L phenylmethylsulfonyl fluoride61. For the histological analyses, embedded fragments were cryosectioned (8 µm sections), fixed with Histochoice (Amresco, Ohio, USA), dipped in 60% isopropanol and then in 0.4% Oil Red O in (60%) isopropanol (for 20 min) to stain for neutral lipids (Oil red O). Vascular smooth muscle cells (alpha-actin) were stained using a primary antibody monoclonal mouse anti-human smooth muscle actin antibody, clone 1A4 (DakoCytomation, Glostrup, Denmark), diluted 1:50 in 10% rabbit serum, and a secondary antibody biotin rabbit anti-mouse Ig (DakoCytomation, Glostrup, Denmark), diluted 1:200 in 10% of rabbit serum. Hemorrhage (glycophorin A) was assessed using a primary antibody monoclonal mouse anti-human glycophorin A(CD235a) (DakoCytomation, Glostrup, Denmark), diluted 1:400 in 10% rabbit serum, and a secondary antibody biotin rabbit anti-mouse F(ab´)2 (DakoCytomation, Glostrup, Denmark), diluted 1:200 in 10% of rabbit serum and CD68 (macrophages) was measured using a primary monoclonal antibody mouse anti-human CD68, clone KP1 (DakoCytomation, Glostrup, Denmark), diluted 1:100 in 10% rabbit serum, and secondary antibody biotinylated polyclonal Rabbit anti-mouse, Rabbit F(ab’)2 (DakoCytomation, Glostrup, Denmark), diluted 1:200 in 10% of rabbit serum. Russell‐Movat pentachrome was used to detect plaque collagens. Sections were scanned using a ScanScope Console Version 8.2 (LRI imaging AB, Vista CA, USA) and photographed with Aperio image scope v.8.0 (Aperio, Vista California, USA). The positively stained plaque area was quantified (blindly) using BiopixiQ 2.1.8 (Gothenburg, Sweden). The histological vulnerability index was calculated as the sum of CD68, neutral lipids (Oil red O) and hemorrhage (glycophorin A) stained plaque areas divided by the sum of smooth muscle cell (alpha-actin) and collagen (MOVAT pentachrome) stained areas.
Growth factors, collagen and matrix metalloproteinase measurements
Sixteen growth factors, all with important roles in cardiovascular disease, were assessed in 218 plaque tissue homogenates, of which 71 patients had T2D. Hepatocyte growth factor, β-nerve growth factor, placental growth factor, vascular endothelial growth factor, platelet-derived growth factor subunit B, stem cell factor, heparin-binding EGF like growth factor, growth differentiation factor, colony stimulating factor-1, fibroblast growth factor, epidermal growth factor and growth hormone were measured in plaque tissue homogenates using a Proximity Extension Assay (PEA; Proseek Multiplex CVD96x96 reagents kit, Olink Bioscience, Uppsala, Sweden) at the Clinical Biomarkers Facility, Science for Life Laboratory, Uppsala. Data were pre-processed for normalization using Olink Wizard for GenEx (Multid Analyses, Sweden). All data are expressed as arbitrary units. Approximate concentrations of growth factors can be calculated from general calibrator curves available at Olink homepage (http://www.olink.com). Platelet derived growth factor-AB/BB and vascular endothelial growth factor were measured using a human Cytokine/chemokine immunoassay (Millipore Corporation, MA, USA) and analysed with Luminex 100 IS 2.3 (Austin, Texas, USA). Absolute concentrations of TGF-ßs (-β1, -β2 and –β3) were measured using Milliplex Map TGF-β Magnetic Bead 3 Plex Kit (MerckMillipore; cat: TGF-ß MAG-64K-03, Billerica, MA, USA) in non-acidified plaque tissue homogenates to ensure that only free/active TGF-ß levels were assessed. Plaque levels of growth factors were normalized to plaque wet weight.
Plaque collagen (acid and pepsin soluble collagens types I to V) content was measured in plaque tissue homogenate using Sircol soluble collagen assay (Biocolor, Carrickfergus, Northern Ireland, UK). The assay detects acid and pepsin soluble collagens types I to V. Results were expressed as collagen levels normalized to plaque wet weight. MMP2 and -9 levels were analyzed in plaque tissue homogenate using Mesoscale human MMP ultra-sensitive kit (Mesoscale, Gaithersburg, MD, USA) as per the provider’s guide. Results were normalized to plaque wet weight.
Carotid plaque bulk RNA sequencing
Bulk RNA sequencing data of 22 human carotid plaques from patients with T2D were used to quantify the expression of TGFB1, TGFB2, TGFB3 and other VSMC marker genes. Total RNA was extracted from the most stenotic plaque region by standard trizol method and cleaned for Ribosomal RNA using Ribo-Zero™ Magnetic Kit from (Epicentre). Strand-specific RNAseq libraries were prepared using ScriptSeq™ v2 RNA-Seq Library v2 Preparation Kit (Epicentre) and sequenced using high-output kit version 2 on HiSeq2000 and NextSeq platform, Illumina, USA. Reads were aligned to human genome assembly GRCh38 using STAR aligner and quantified by Salmon with gene annotation GENCODE V2762,63. Counts were normalized by edgeR, and gene expression was normalized as log2- transformed count per million (CPM)64. Subsequently, batch effects of sequencing platforms (Illumina HiSeq2000 and the NextSeq platforms) were adjusted by an empirical Bayes method Combat65. The batch-corrected log2CPM was used for further analysis.
Carotid plaque single-cell RNA sequencing
Carotid plaque cells were isolated using a previously published enzymatic dispersion method with some modifications66. Plaque tissue was taken immediately after surgical removal and placed in RPMI 1640 media. To remove circulating cells and red blood cells, plaques were washed with RPMI, and red blood cells were lysed using red blood cell lysis buffer (Red Blood Cell Lysing Buffer Hybri-Max™, Sigma-Aldrich). Subsequently, the plaque tissue was cleaned of calcified areas, minced and placed into an enzymatic digestion cocktail consisting of collagenase type I (400 units/mL, Sigma C9722), elastase type III (5 units/mL, Worthington, LS006365), and DNase (300 units/mL, Sigma D5025), with 1 mg/mL soybean trypsin inhibitor (Sigma T6522), 2.5 μg/mL polymixin B (Sigma), and 2 mM CaCl2, in RPMI medium 1640 with 5% FBS. The suspension was incubated at 37 °C for 30 min with continuous agitation. After incubation, cell suspension was pipetted up and down to break the remaining tissue. Thereafter, the cell suspension was strained using a 100 µm strainer and pelleted by centrifugation at 500×g for 5 min. The cells were then suspended in fresh RPMI 1640 media. For sorting purpose, isolated cells were washed, stained with MitoTracker™ Green FM (ThermoFisher Scientific, Cat# M7514) (50 nM 30 min at RT), FC blocked (Biolegend, Cat# 422302) (1:33 15 min at RT), and subsequently incubated with an antibody cocktail consisting of PE/Cy7 anti-human CD235a (Glycophorin A, Biolegend, Cat# 349111), and APC anti-human CD45 (Biolegend, Cat# 304012) antibodies at 1:100 dilution for 30 min at 4 °C. Thereafter, cells were washed, resuspended in PBS containing viability dye 7-AAD (Biolegend, Cat# 420403) at 1:100 dilution and processed immediately for FACS sorting using a BD FACSAria III cell sorter (BD Biosciences). CD45+ and CD45− cells were separately sorted into 384 wells of Smart seq2 plates (Eukaryotic genomic facility, Scilife lab, Stockholm) following user guidelines. The sorted plates were frozen instantly on dry ice and delivered to Scilife lab for sequencing.
Quality check, library preparation and sequencing were performed according to standard methods at Scilife eukaryotic genomic facility according to a published protocol, same as the processing of the raw single-cell RNA sequencing data67. Cells were dropped for low-quality libraries less than 10,000 raw reads. Cells were further filtered by the Seurat if the number of detected genes was less than 500 and the percentage of ERCC RNA Spike-In was no more than 15%68. Counts were log-normalized, scaled and top 2000 highly variable genes were used for dimensional reduction. As determined by the percentages of variance explained by principal components (ElbowPlot functions of the Seurat69), the first seven principal components were used to construct a shared nearest neighbor graph and a non-linear dimensional reduction t-distributed stochastic neighbor embedding (t-SNE) for CD45+ and CD45− cells, respectively. Cell clusters were then identified by the default Louvain algorithm using “FindClusters” function of the Seurat (resolution = 0.5).
Cell type per cluster was determined by canonical cell markers. Cell types were also predicted using single cell reference data of human vasculature from the Tabula68 by the “TransferData” function of Seurat69.
Genes highly expressed in one cluster compared to all other clusters were examined by two-sided Wilcoxon Rank-Sum test using the “FindMarkers” function of Seurat. Genes with an average log-2 transformed fold change above 0.25 and adjusted p-value less than 0.05 were considered as differentially expressed genes (DEG). Consequently, an R package ReactomePA was implemented to perform pathway enrichment analysis using these DEGs. A significant enriched pathway had a p-value less than 0.05 after Benjamini–Hochberg correction. Module scores for the selected pathway were calculated using “AddModuleScore” function from the Seurat R package (version 3.2.3).
Validation of the identified cell types in an independent dataset
Publicly accessible single-cell RNA sequencing (scRNA-seq) data from human carotid plaques70, encompassing both the atherosclerotic core (AC) and the proximal adjacent (PA) region, were utilized to validate our findings. This independent dataset was analyzed through a reproducible pipeline designed for scRNA-seq71. Based on gene expression of markers for vascular smooth muscle cells (VSMCs, Supplementary Fig. 7), the cells from clusters 5, 8, 11 and 12 (obtained from the PlaqView pipeline71) were suggested as VSMCs in the independent dataset. Focusing on these VSMCs, we examined the expression of main differentially expressed genes (DEGs) of the identified VSMCs clusters (namely contractile, adipocyte-like, synthetic/fibroblast-like and macrophage-like VSMCs) from our dataset (Supplementary Fig. 8). Additionally, we predicted VSMC cell types by using our CD45− cell types as reference. The prediction score for each cell was obtained by label transfer function of the Seurat (Supplementary Fig. 8).
Gene expression of markers for immune cells such as T-cells (CD3E, CD4, CD8A), NK cells (NCAM1, NKG7, XCL1), myeloid cells (CD163, FCGR1A, ITGAX, C1QA, IL1B, CD1C), B-cells (CD79A), plasmacytoid dendritic cells (CLEC4C) and mast cells (KIT) were examined in this independent data. Based on gene expression of these markers, cells in clusters 0, 1 and 17 (obtained from the PlaqView pipeline) were annotated as T-cells, cluster 6 as NK cells, clusters 3, 4 and 7 as myeloid cells, clusters 10 and 14 as B cells, cluster 18 as plasmacytoid dendritic cells and cluster 16 as mast cells. Gene expression of markers for non-immune cells (VSMC: ACTA2, MYH11, TAGLN; fibroblasts: PLA2G2A, FBLN1; endothelial cells: PECAM1, VWF) were also examined. As previously described, cells from clusters 5, 8, 11 and 12 were annotated as VSMCs, cluster 13 as fibroblast, clusters 2, 9 and 15 as endothelial cells. Next, gene expression of TGFB isoforms was examined in this independent data, split by immune cells (T, NK, Myeloid, pDC, mast) and non-immune cells (VSMC, fibroblast, endothelial cells; Supplementary Fig. 10).
Validation of the TGF isoform expression in an independent dataset
Using the same dataset as in the cell type validation, the expression of the three TGFB isoforms was assessed. When comparing the TGFB2 expression between the Smart-Seq2 (SS2) single-cell analysis to the 10x single-cell RNA sequencing analysis, we identified that TGFB2 expression was detected in higher levels by SS2 than by the 10x (p = 2.2 × 10−32). We compared the TGFB2 expression between the core and proximal regions and found that TGFB2 was detected to a greater extent in proximal regions (Supplementary Fig. 9a). These were then used to validate the expression of TGFB2 among the VSMCs. Considering the very low expression of TGFB isoforms and heterogeneity of plaques, negative binomial regressions were applied to compare the expression of TGFB2 between the predicted contractile VSMCs and the rest of the VSMCs within the proximal region for each patient. A fixed-effect meta-analysis employing the inverse-variance method was performed to aggregate the differences in expression between contractile VSMCs and the other VSMCs within proximal regions. Results are shown in Supplementary Fig. 9b.
Visium spatial transcriptomics of carotid plaques
Plaque sections for Visium spatial transcriptomics were taken from the most stenotic region of the plaques and processed according to the user’s guide (CG000239 RevD). OCT embedded plaque sections (10 µm) were placed on Visium tissue sample slides, fixed with methanol, stained with hematoxylin and eosin, and imaged by ScanScope Console Version 8.2 (LRI imaging AB, Vista CA, USA). Thereafter, samples were permeabilized for 12 min and libraries were prepared according to the Visium Spatial Gene Expression protocol. Library quality was assayed using a Bioanalyzer High Sensitivity chip (Agilent) and 2.0pM of library was sequenced on NextSeq 500/550 using high Output Kit v2.5 (150 Cycles) at a sequencing depth of 400 million reads-pair per sample. Sequencing was performed using the following read protocol: read 1: 28 cycles; i7 index read: 10 cycles; i5 index read: 10 cycles; and read 2: 91 cycles.
Raw data for each plaque (FASTQ files) and respective histological images were processed with the Space Ranger software v.1.0.0, which uses STAR v.2.5.1b52 for genome alignment, against the Cell Ranger hg38 reference genome refdata-cellranger-GRCh38-3.0.0, available at: http://cf.10xgenomics.com/supp/cell-exp/refdata-cellranger-GRCh38-3.0.0.tar.gz. Further analyses were done by using the Seurat. Mitochondria DNA encoded genes were removed prior to analysis. After that, low-quality spots were filtered if the number of the detected genes was less than 50. Counts were normalized by a regularized negative binomial model sctransform which accounted for technical artifacts while preserving biological variance. Using an ‘anchor’-based integration and all the obtained clusters of CD45+ and CD45− cells from our single-cell RNAseq analysis as references, prediction scores for each spot for each class of CD45+ and CD45− cell types were obtained by label transfer of the Seurat, respectively. Cell type corresponding to the top prediction score per spot was assigned to the spot.
In vitro studies of smooth muscle cell differentiation and wound healing
Human coronary artery smooth muscle cells (HCASMC) were obtained from ThermoFisher Scientific (Cat# C0175C) and cultured in Human Vascular Smooth Muscle Cell Basal Medium supplemented (Cat# M231500, ThermoFisher Scientific) with smooth muscle cell growth supplement (Cat# S00725, ThermoFisher Scientific), 100 U/mL penicillin and 100 μg/mL streptomycin in CO2 incubator at 37 °C. For comparison of genes between two phenotypes, 5 × 105 proliferating HCASMS were grown to 80% confluency in a 12-well plate and differentiated by transferring into differentiation media (Cat# S0085, ThermoFisher Scientific) as recommended. Following 6 days of differentiation, cells were harvested for mRNA isolation using Qiagen RNeasy kit (Qiagen, Cat# 74106) according to the manufacturer’s instructions. For comparisons, mRNA from proliferative HCASMC grown in parallel was isolated.
To study if differentiated HCASMCs induce a phenotypic shift in proliferative HCASMCs two different approaches were used. First, 5 × 105 proliferating HCASMS were exposed to wasted media from 6 days differentiated contractile HCASMCs for 72 h before cells were harvested and mRNA was isolated. In an alternate and more direct approach, proliferative HCASMC were exposed to 5 ng/mL TGF-ß2 for up to 72 h before cells were harvested for mRNA isolation.
In a separate series of experiments, we studied the effect of hyperglycemic conditions on proliferative HCASMCs migration using a wound healing assay. In brief, 1 × 104 proliferating HCASMCs were seeded in 24-well plates for 48 h. Cells were serum-deprived for 24 h and a scratch (in the middle of each well) was created in the cell monolayer. Cells were then washed and incubated with fresh basal growth media containing 4.6 or 20 mM glucose. To acquire images from the same field across different time points, a reference marking was made by etching the outer bottom of each plate with a razor blade. The images were acquired at 0, 8, 12 and 16 h post glucose stimulation using a Nikon phase contrast (DS-Fi1) microscope using a 10X objective and NIS-Elements Basic Research software. The percentage of wound closure was calculated on image areas analyzed by MRI-wound healing plug-in in ImageJ software (NIH, USA), using Eq. 1. Data was summarized with images from 8 wells.
$${Wound\; closue}\, \left(\%\right)=100\%\,*\, \left(1-{{{{\rm{A}}}}}_{{{{{\rm{T}}}}}_{{{{\rm{x}}}}}}/{{{{\rm{A}}}}}_{{{{{\rm{T}}}}}_{0}}\right)$$
(1)
where \({{{{\rm{A}}}}}_{{{{{\rm{T}}}}}_{{{{\rm{x}}}}}}\) and \({{{{\rm{A}}}}}_{{{{{\rm{T}}}}}_{0}}\) represented wound areas at respective time points and 0 h post glucose stimulation.
Quantitative PCR
Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Cat# 74106), according to the manufacturer’s instructions. Then, 1 μg of total RNA was reverse-transcribed using a High capacity RNA-to-cDNA kit (ThermoFisher Scientific, Cat# 4387406) as per user guidelines. Gene expressions were examined by quantitative real-time PCR on a QuantStudio 7 Flex instrument (Applied Biosystems/ThermoFisher) using Taqman Fast Advanced master mix (ThermoFisher Scientific, Cat# 4444557) and appropriate Taqman probes (Supplementary Table 9). Relative gene expression was calculated with QuantStudio Software v1.1 (ThermoFisher) using the ΔΔCt method and normalized to GAPDH expression as endogenous control. For comparison of different groups, gene expression levels are expressed as fold change expression compared to control samples.
Western blotting
Cultured proliferative HCASMCs were lysed directly in 1x Laemmli sample buffer (#1610747, Bio-Rad) and boiled 10 min at 95 °C under intensive shaking to shear DNA. Proteins were separated by SDS-PAGE (using 4–15% Mini-PROTEAN® Criterion TGX Stain-Free gels; #456-8084, Bio-Rad) and transferred to PVDF (Polyvinylidene fluoride) membranes (Bio-Rad). The membrane was blocked with 5% bovine serum albumin in TBST (20 mM Tris, 150 mM NaCl, 0.1% Tween) and incubated overnight at 4 °C with primary Anti-p-SMAD3 (phospho S423+S425; 1:1000, # ab52903, Abcam) or Anti-p-SMAD2 (phospho S467; 1:1000, #ab53100, Abcam). On the following day, the PVDF membrane was washed with TBST buffer 5 times (5 min each), incubated with anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibodies (1:1000, #P0448, Dako) and kept at room temperature for 1 h. The membrane was washed with TBST buffer as before. Enhanced chemiluminescent (ECL) substrates (#32106, ThermoFisher) were employed for the detection of HRP enzyme activity.
For re-probing, the antibodies were stripped using a western blot stripping solution (#21059, ThermoFisher) for 10 min. Thereafter, membranes were blocked with 5% bovine serum albumin in TBST and incubated overnight at 4 °C with primary Anti-SMAD3 (1:1000, # ab40854, Abcam) or Anti-SMAD2 (1:1000, # ab40855). On the following day, the PVDF membrane was washed, incubated with anti-rabbit (HRP)-conjugated secondary antibodies for 1 h, washed with TBST as before, and ECL substrates were employed for the detection of HRP enzyme activity. Then the antibodies were stripped again and the PVDF membrane was incubated with Anti-GAPDH (1:5000, #ab8245, Abcam) for loading control for 2 h and incubated with anti-mouse (HRP)-conjugated secondary antibodies (1:1000, #P0447, Dako) for 1 h, washed with TBST as before and ECL substrates were employed for the detection of HRP enzyme activity.
Protein bands were visualized on a ChemiDoc MP instrument (Bio-Rad) and subjected to densitometric quantification using ImageLab v6.1 software (Bio-Rad).
Plaque cell experiments
Plaque cells were isolated as described above and cultured in RPMI 1640 media supplemented with 10% FBS and penicillin and streptomycin (1%). After 24 h of incubation, media was collected and centrifuged at 500×g for 5 min to remove debris. The clean supernatant was used for the assessment of TGF-β release as described above.
Zymography of plaque cells to measure MMP activity
To determine MMP2 activity, the supernatant of plaque cells from patients with type 2 diabetes or without diabetes, or the supernatant of HCASMCs stimulated for 48 h with 5 (control), 10, and 20 mM glucose, was first mixed with 2x sample buffer (ThermoFisher Scientific, LC2676) and loaded onto Novex 10% Zymogram Plus (Gelatin) Protein Gels (ThermoFisher Scientific ZY00100BOX) and ran at constant 200 V for 40 min. Afterward, the gel was incubated with 1x Renaturing Buffer (ThermoFisher Scientific, LC2670) for 30 min at room temperature with gentle agitation. The buffer was then replaced with 1x Developing Buffer (ThermoFisher Scientific, LC2671) for 30 min at room temperature, and successively replaced with new 1x Developing Buffer for 24 h at 37 °C. Finally, the gel was rinsed with distilled water and incubated (30 min to 2 h) with Coomassie Brilliant Blue. The areas of protease activity appear as clear bands against a dark background. Bands were visualized on a ChemiDoc MP instrument (Bio-Rad) and subjected to densitometric quantification using ImageJ 1.54f.
Follow-up
(CV) events (including myocardial infarction, cardiovascular death, amaurosis fugax and stroke (ipsilateral and contralateral events)) for T2D patients were retrieved from the Swedish National Inpatient Health Register (2005–2013) during the follow-up (up to 85 months). Loss of follow-up (due to emigration from Sweden) or death due to other causes than cardiovascular were censored. Kaplan–Meier survival analysis was conducted comparing patients with vulnerability index (VI) in 1st and 2nd tertiles to 3rd tertile, as planned a priori to gain power.
Multivariate analysis
Orthogonal partial least squares discriminant analysis (OPLS-DA) was performed in SIMCA-P software package (version 17, Umetrics, Umeå, Sweden). Prior to analysis, data was mean-centered and scaled to unit variance (UV) to remove systematic differences in variables caused by differences in their measurement units. Model performance was evaluated by R2 and Q2 values varying from 0 to 172. R2 represents the proportion of variance in T2D explained by the model, whereas Q2 is a measure of the model’s predictive ability, obtained through 7-fold cross-validation. Variables responsible for separation between classes were examined from the variable influence on projection (VIP) values of each variable. VIP is defined as the fraction to which each variable explains Y variance and ranks the variables according to their contribution to the model. Hierarchical clustering of variables was performed by using the Euclidean distance matrix and average linkage method by using the R package pheatmap (version 1.0.12).
Statistics
Mann–Whitney U and Chi-square tests were used for comparisons between continuous and categorical variables and Spearman’s rho statistics for assessing correlations between continuous variables. Multiple linear regression was used to examine the components of vulnerability index in plaques from T2D and ND subjects and to identify factors associated with plaque levels of free TGF-ß2. Log rank test was used in Kaplan–Meier survival analysis. Orthogonal partial least squares discriminant analysis (OPLS-DA) was performed in SIMCA-P (version 17, Umetrics, Umeå, Sweden). In this analysis, model performances were evaluated by R2 and Q2 values. ANOVA of the cross-validated residuals (CV-ANOVA) was implemented to examine the significance of the OPLS-DA model. Contribution of variables in classes separations were examined by the variable influence on the projection (variable importance plot; VIP) values of each variable. Hierarchical clustering was implemented using the average linkage method and Euclidean distance in R (version 4.2.2). Statistical analyses were performed on SPSS 22.0 (IBM Corp., Armonk, NY, USA) or GraphPad Prism 9 (GraphPad Software, Boston, USA) unless stated otherwise.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.

 Arabic
Arabic Dutch
Dutch English
English French
French German
German Italian
Italian Portuguese
Portuguese Russian
Russian Spanish
Spanish