
Table of Contents
Mice
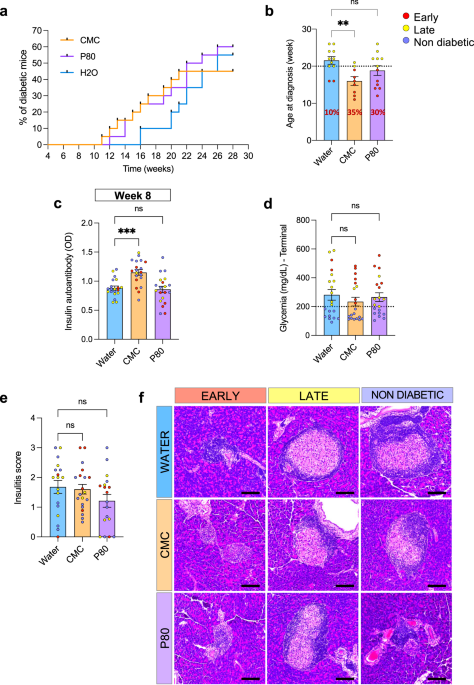
Four-week-old female NOD/ShiLtJ mice were purchased from Jackson Laboratories. Mice were housed in cages of 5 mice and were kept under a 12 h light/dark cycle and had free access to standard chow diet and water (water-treated group, 4 cages) or water with 1% CMC (CMC-treated group, 4 cages) or water with 1% P80 (P80-treated group, 4 cages). Cages from all groups were changed every other week. Body weight was measured and feces were collected every other week. From week 8 of age, glycemia was checked every 2 weeks and every week from week 12 of age. Serum was collected at week 8 of age. Type one diabetes status was declared after 2 glycemia measurements above 200 mg dL−1 72 h apart. Upon diabetes diagnosis, or after 28 weeks of age, mice were euthanized by isoflurane and cervical dislocation. Colon length, colon weight, spleen weight, and adipose weight were measured and organs were collected for downstream analysis. Animal welfare and experimental protocols followed the ARRIVE guidelines (Animal Research: Reporting of In Vivo Experiments). All procedures involving animals were approved by the French Ministère de la l’enseignement supérieur, de la recherche et de l’innovation, APAFIS#24788-2019102806256593 v8.
Quantification of serum insulin auto-antibody by ELISA
Insulin-specific serum IgG levels were quantified by ELISA. Microtiter plates were coated overnight with purified mouse insulin (100 ng per well). Serum samples diluted 1:40 were then applied. After incubation and washing, wells were incubated with HRP-linked anti-mouse IgG (1:1000, SouthernBiotech, 1015-05). Quantification was performed using the colorimetric peroxidase substrate tetramethylbenzidine and optical density was read at 450 nm (Versamax microplate reader). Data are reported as optical density corrected by subtracting background (determined by readings in samples lacking serum).
Quantification of serum flagellin or LPS-specific IgG by ELISA
Flagellin and LPS-specific serum IgG levels were quantified by ELISA. Microtiter plates were coated overnight with purified flagellin from Salmonella Typhimurium (Sigma, 100 ng per well) or LPS (from E. coli 0128: B12, Sigma, 2 μg per well) diluted in carbonate–bicarbonate buffer. Serum samples diluted 1:200 were then applied. After incubation and washing, wells were incubated with HRP-linked anti-mouse IgG (1:1000, SouthernBiotech, 1015-05). Quantification was performed using the colorimetric peroxidase substrate tetramethylbenzidine and optical density was read at 450 nm (Versamax microplate reader). Data are reported as optical density corrected by subtracting background (determined by readings in samples lacking serum).
Quantification of fecal LCN2 by ELISA
For quantification of fecal LCN2 by ELISA, frozen fecal samples were reconstituted in PBS containing 0.1%Tween 20 to a final concentration of 100 mg ml−1 and vortexed for 20 min to produce a homogenous fecal suspension29. These samples were then centrifuged for 10 min at 14,000×g and 4 °C. Clear supernatants were collected and stored at −20 °C until analysis. LCN2 levels were estimated in the supernatants using a Duoset murine LCN2 ELISA kit (R&D Systems, Minneapolis, MN, USA) using the colorimetric peroxidase substrate tetramethylbenzidine, and optical density was read at 450 nm (Versamax microplate reader).
Insulinitis scoring
Following euthanasia, pancreases were harvested and fixed in 4% paraformaldehyde, embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Insulitis was evaluated blindly and as previously described24. Briefly, all islets on the slide were analyzed and a score between 0 and 3 was given to each. 0 = no insulitis, 1 = peri-insulitis, 2 = infiltration < 50% of the islet and 3 = infiltration > 50% of the islet.
Hematoxylin and eosin staining and histopathologic analysis
Following euthanasia, colons (proximal colon, 2 first cm from the cecum) were placed in Carnoy’s fixative solution (60% methanol, 30% chloroform, 10% glacial acetic acid). Tissues were then washed in methanol 2 × 30 min, ethanol 2 × 15 min, ethanol/xylene (1:1) 15 min, and xylene 2 × 15 min, followed by embedding in paraffin with a vertical orientation. Tissues were sectioned at 5-μm thickness and stained with hematoxylin & eosin (H&E) using standard protocols. H&E-stained slides were assigned four scores based on the degree of epithelial damage and inflammatory infiltrate in the mucosa, submucosa, and muscularis/serosa. Each of the four scores was multiplied by 1 if the change was focal, 2 if it was patchy, and 3 if it was diffuse, as previously described29. The four individual scores per colon were added, resulting in a total scoring range of 0–36 per mouse.
Microbiota analysis by 16S rRNA gene sequencing
16S rRNA gene amplification and sequencing were done using the Illumina MiSeq technology following the protocol of Earth Microbiome Project with their modifications to the MOBIO PowerSoil DNA Isolation Kit procedure for extracting DNA (www.earthmicrobiome.org/emp-standard-protocols). Bulk DNA were extracted from frozen extruded feces using a PowerSoil-htp kit from MoBio Laboratories (Carlsbad, CA, USA) with mechanical disruption (bead-beating). The 16S rRNA genes, region V4, were PCR amplified from each sample using a composite forward primer and a reverse primer containing a unique 12-base barcode, designed using the Golay error-correcting scheme, which was used to tag PCR products from respective samples45. We used the forward primer 515F 5’- AATGATACGGCGACCACCGAGATCTACACGCTXXXXXXXXXXXXTATGGTAATTGTGTGYCAGCMGCCGCGGTAA-3’: the italicized sequence is the 5’ Illumina adapter, the 12 X sequence is the golay barcode, the bold sequence is the primer pad, the italicized and bold sequence is the primer linker and the underlined sequence is the conserved bacterial primer 515F. The reverse primer 806R used was 5’-CAAGCAGAAGACGGCATACGAGATAGTCAGCCAGCC GGACTACNVGGGTWTCTAAT-3’: the italicized sequence is the 3’ reverse complement sequence of Illumina adapter, the bold sequence is the primer pad, the italicized and bold sequence is the primer linker and the underlined sequence is the conserved bacterial primer 806R. PCR reactions consisted of Hot Master PCR mix (Quantabio, Beverly, MA, USA), 0.2 μM of each primer, 10–100 ng template, and reaction conditions were 3 min at 95 °C, followed by 30 cycles of 45 s at 95 °C, 60 s at 50 °C, and 90 s at 72 °C on a Biorad thermocycler. Products were then visualized by gel electrophoresis and quantified using Quant-iT PicoGreen dsDNA assay (Clariostar Fluorescence Spectrophotometer). A master DNA pool was generated in equimolar ratios, subsequently purified with Ampure magnetic purification beads (Agencourt, Brea, CA, USA) and sequenced using an Illumina MiSeq sequencer (paired-end reads, 2 × 250 bp) at the Genom’IC platform (INSERM U1016, Paris, France).
16S rRNA gene sequence analysis
16S rRNA sequences were analyzed using QIIME2—version 201946. Sequences were demultiplexed and quality filtered using the Dada2 method47 with QIIME2 default parameters in order to detect and correct Illumina amplicon sequence data, and a table of QIIME2 artifact was generated using the following dada2 command: qiime dada2 denoise-paired –i-demultiplexed-seqs demux.qza –p-trim-left-f 0 –p-trim-left-r 0 –p-trunc-len-f 180 –p-trunc-len-r 180 –o-representative-sequences rep-seqs-dada2.qza –o-table table-dada2.qza –o-denoising-stats stats-dada2.qza –p-n-threads 6. A tree was next generated, using the align-to-tree- mafft-fasttree command, for phylogenetic diversity analyses, and alpha and beta diversity analyses were computed using the core-metrics-phylogenetic command. Principal coordinate analysis (PCoA) plots were used to assess the variation between the experimental group (beta diversity). For taxonomy analysis, features were assigned to operational taxonomic units (OTUs) with a 99% threshold of pairwise identity to the SILVA reference database48.
Fecal flagellin, LPS, and TLR2 ligands load quantification
Levels of fecal bioactive flagellin, LPS, and TLR2 ligands were quantified as previously described49 using human embryonic kidney (HEK)-Blue-mTLR5, HEK-Blue-mTLR4, and HEK-Blue-mTLR2 cells, respectively (Invivogen, San Diego, CA). We resuspended fecal material in PBS to a final concentration of 100 mg mL−1 and homogenized for 15 minutes using a vortex. We then centrifuged the samples at 8000×g for 15 min and serially diluted the resulting supernatant and applied it to mammalian cells. Purified Escherichia coli flagellin, LPS (Sigma, St. Louis, MO, USA), and TLR2 ligands were used for standard curve determination using HEK-Blue-mTLR5, HEK-Blue-mTLR4 and HEK-Blue-mTLR2 cells, respectively. After 24 h of stimulation, we applied cell culture supernatant to QUANTI-Blue medium (Invivogen, San Diego, CA, USA) and measured alkaline phosphatase activity at 620 nm after 30 min.
Immunostaining of mucins and localization of bacteria by fluorescent in situ hybridization
Mucus immunostaining was paired with fluorescent in situ hybridization (FISH), as previously described14,50, in order to analyze bacteria localization at the surface of the intestinal mucosa. In brief, colonic tissues (proximal colon, second cm from the cecum) containing fecal material were placed in methanol-Carnoy’s fixative solution (60% methanol, 30% chloroform, 10% glacial acetic acid) for a minimum of 3 h at room temperature. Tissues were then washed in methanol 2 × 30 min, ethanol 2 × 15 min, ethanol/xylene (1:1) 15 min, and xylene 2 × 15 min, followed by embedding in paraffin with a vertical orientation. Five-μm sections were cut and dewaxed by preheating at 60 °C for 10 min, followed by bathing in xylene at 60 °C for 10 min, xylene at room temperature for 10 min, and 99.5% ethanol for 10 min. The hybridization step was performed at 50 °C overnight with an EUB338 probe (59-GCTGCCTCCCGTAGGAGT-39, with a 59 Alexa 647 label) diluted to a final concentration of 10 mg mL−1 in hybridization buffer (20 mM Tris–HCl, pH 7.4, 0.9 M NaCl, 0.1% SDS, 20% formamide). After washing for 10 min in wash buffer (20 mM Tris–HCl, pH 7.4, 0.9 M NaCl) and 3×10 min in PBS, a PAP pen (Sigma, St. Louis, MO, USA) was used to mark around the section and block solution (5% FBS in PBS) was added for 30 min at 4 °C. Mucin-2 primary antibody (rabbit H-300, [C3], C-term, Genetex, GTX100664) was diluted to 1:100 in block solution and applied overnight at 4 °C. After washing 3 × 10 min in PBS, block solution containing anti-rabbit Alexa 488 secondary antibody diluted to 1:300, PhalloidinTetramethylrhodamine B isothiocyanate (Sigma-Aldrich) at 1 mg ml−1 and Hoechst 33258 (Sigma-Aldrich) at 10 mg ml−1 was applied to the section for 2 h. After washing 3 × 10 min in PBS slides were mounted using Prolong anti-fade mounting media (Life Technologies) and kept in the dark at 4 °C. Observations and measurement of the distance between bacteria and epithelial cell monolayer were performed with a Spinning Disk IXplore using the Olympus cellSens imaging software 421 (V2.3) at a frame size of 2048 × 2048 with 16-bit depth. A 405 nm laser was used to excite the 422 Hoechst stain (epithelial DNA), 488 nm for Alexa Fluor 488 (mucus), 488 nm for Phalloidin (actin), 423 and 640 nm for Alexa Fluor 647 (bacteria). Samples were imaged with a ×20 objective.
Quantification of fecal IgA-coated bacteria and bacterial load
IgA-coated bacteria were quantified as previously described28. In brief, frozen fecal samples were thoroughly homogenized in PBS to a final concentration of 20 mg/ml. Fecal suspensions were filtered through a 40-μm sterile nylon mesh, then centrifuged at 50×g, for 15 min at 4 °C. 200 μl of supernatant was then washed with 1 ml PBS and centrifuged at 8000×g, for 5 min at 4 °C. Resulting bacterial pellets were resuspended in 100 μl blocking buffer (staining buffer containing 20% Normal Rat Serum) and incubated for 20 min on ice before being stained with 100 μl of staining buffer containing PE-conjugated Anti-Mouse IgA (1:12.5; eBioscience, 12-4204-82) for 30 min on ice, in the dark. Following two washes with staining buffer, pellets were resuspended in 200 μl of FACS buffer (PBS, 1% Normal Rat Serum). Data acquisition was performed on a Beckman Coulter Gallios flow cytometer. For each sample, 50,000 events were recorded and data was analyzed using FlowJo software v.10.8.2. Bacterial load was quantified by gating bacterial population based on size and granularity.
Colonic RNA extraction and q-RT-PCR analysis
Distal colon was collected during euthanasia and placed in RNAlater. Total mRNAs were isolated from colonic tissues using TRIzol (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions and as previously described29. Quantitative RT-PCR was performed using the Qiagen kit QuantiFast® SYBR® Green RT-PCR in a LigthCycler® 480 instrument (Roche Molecular Systems, Inc.) with specific mouse oligonucleotides (Supplementary Table 1). Gene expressions are presented as relative values using the Ct approach with the Gapdh housekeeping gene.
Treatment and diabetic status prediction based on microbiota composition
The association between microbiota composition and treatment or diabetic status was assessed using prediction based on microbiota composition or pro-inflammatory potential data of either CMC treatment (outcome CMC or WATER, Fig. 6a, e, i, m), P80 treatment (outcome P80 or WATER, Fig. 6b, f, j, m), diabetic status (outcome TRUE or FALSE, Fig. 6c, g, k, o) or early diabetic status (outcome EARLY or LATE-OR-NO, Fig. 6d, h, l, p). Receiver operating characteristic (ROC) curves were calculated (R version 4.1.2, randomForest 4.7-1.1 package, ROCR package) using training data set and validation data set containing randomly affected 80% and 20% of mice, respectively. Data set contained relative abundance data for microbiota members identified at the species level at week 4 (Fig. 6a–d), at week 8 (Fig. 6e–h), at week 10 (Fig. 6i–l) or quantitative microbiota functional features (fecal levels of flagellin, LPS, TLR2 ligands at weeks 8 and 10, distance from intestinal epithelial cells, week 10 fecal proportion of IgA-coated bacteria, and flagellin/LPS specific antibodies levels at week 8 of age and euthanasia, Fig. 6m–p). ROC calculation was repeated 20 times with random sampling of the training and validation data and area under curve (AUC) measurement for each iteration. Mean AUC and standard deviation are presented for each graph.
Identification of microbiota members significantly altered in their relative abundance
Microbiota members presenting significant changes in abundance after 6 weeks of CMC or P80 treatment were identified using MaAsLin2 (Microbiome Multivariable Associations with Linear Models, version 2)51. MaAsLin2 analysis (R version 4.1.2, Maaslin2 version 1.12.0 package) was conducted using relative abundance data for microbiota members identified at the species level at week 10. Microbiota members were reported as significantly altered in their relative abundance if corrected p-value < 0.05. Obtained data were reported as a Log2 fold change in relative abundance compared to the mean abundance observed in the Water-treated group.
Statistical analysis
Significance was determined using a log-rank Mandel–Cox test in Fig. 1a. When normality and homoscedasticity postulates were valid, significance was tested using one-way group analysis of variance (ANOVA) with Sidak’s multiple comparisons test (Figs. 1c–e; 3j; 4c, d, g; 5a, b, e, j; Supplementary Figs. 2, 3). Significance of data that did not respect normality and homoscedasticity postulates was tested using Kruskal–Wallis corrected for multiple comparisons with a Dunn’s test (Figs. 1b; 4a, f; 5d, f–i) or Brown–Forsythe and Welch ANOVA corrected for multiple comparisons with a Dunnett test (Fig. 4e), respectively. Significance of longitudinally measured data was assessed using two-way ANOVA corrected for multiple comparisons with Sidak’s test (Figs. 2e, f; 3a–i; Supplementary Fig. 4). Clustering significance in Fig. 2a–d was determined using Permutational multivariate analysis of variance (PERMANOVA) and corrected p-values are indicated on plots. Differences were noted as significant *p ≤ 0,05; **p ≤ 0,01; ***p ≤ 0,001; ****p ≤ 0,0001; n.s. indicates nonsignificant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
